Résumé
Le domaine relativement nouveau de la génomique comparative, dont le développement est rapide, apporte quantité de données utiles à l’évaluation de l’hypothèse que les êtres humains partagent une ascendance commune avec d’autres formes de vie. De nombreux éléments de preuve génomiques indépendants soutiennent fortement l’hypothèse que notre espèce partage un ancêtre commun avec d’autres primates. De plus, d’autres données indiquent aussi que notre espèce a maintenu une taille de population d’au moins plusieurs milliers depuis notre spéciation d’avec les ancêtres d’autres grands singes. Cet article présente une vue d’ensemble des preuves génomiques d’une ascendance commune et des tailles de population d’hominidés, ainsi qu’une discussion des implications de ces éléments de preuve pour les approches scientifiques concordistes aux narratives de la Genèse.
La Genèse et le génome : éléments de preuve génomiques d’un ancêtre commun à l’être humain et au singe et taille des populations ancestrales d’hominidés
La théorie évolutionnaire propose depuis longtemps que les humains et autres grands singes partagent des ancêtres communs.1 La théorie évolutionnaire prédit donc que les génomes que nous observons chez les primates vivants (tels que les humains et les chimpanzés) sont, en réalité, des formes modifiées d’un génome original présent chez l’ancêtre commun de ces espèces. Cette simple hypothèse peut être aisément testée à partir de plusieurs pistes indépendantes dérivées de la comparaison des génomes complets des deux espèces.2
Le premier élément de preuve, qui est peut-être un des plus abordés par les organismes d’apologétique chrétienne, est celui de la similarité des séquences génétiques. Si, effectivement, les humains et les chimpanzés descendent d’une espèce ancestrale commune, alors les séquences génétiques individuelles de ces deux espèces devraient exhiber un haut degré de similarité en raison de l’hérédité d’un ancêtre commun, c’est-à-dire de l’homologie. De plus, il devrait exister une homologie de gènes individuels à deux niveaux : au niveau des acides aminés (la séquence fonctionnelle de la protéine dérivée d’un gène donné), et au niveau du code des nucléotides (le code ADN sous-jacent de la séquence d’acides aminés requise). Etant donné que le code de nucléotides possède de nombreuses options de codage pour une séquence donnée d’acides aminés (c’est-à-dire que le code des nucléotides est redondant), les gènes d’organismes apparentés devraient non seulement partager des séquences d’acides aminés mais aussi des séquences de nucléotides, en dépit du grand nombre d’options de codage possible.
La synténie constitue un deuxième élément de preuve distinct. Ce terme technique décrit la conservation de l’ordre des gènes sur les chromosomes entre les membres d’une famille. Plus simplement, l’hypothèse d’une ascendance commune prédit non seulement que les espèces apparentées possèderont des gènes similaires, mais encore que ces gènes seront disposés de manière très similaire dans l’espace.
Les pseudogènes constituent un troisième élément de preuve. Les pseudogènes (littéralement, « faux gènes ») sont les vestiges mutés de séquences géniques qui persistent dans le génome après leur inactivation. L’ascendance commune prédit que les espèces apparentées devraient partager des pseudogènes présents dans le génome de leur ancêtre commun. De plus, ces pseudogènes devraient se trouver aux mêmes endroits dans le génome des deux espèces descendantes (c’est-à-dire qu’ils devraient démontrer une synténie commune) et garder une similarité de séquence génique (c’est-à-dire qu’ils devraient continuer à montrer une homologie) malgré leur inactivation.
La séquence ADN du génome humain a été terminée et publiée entre 2001 et 20043, suivie peu de temps après par celle du chimpanzé4. La disponibilité de séquences génomiques complètes pour ces deux organismes permet une comparaison d’homologie, de synténie et de pseudogènes partagés au niveau du génome entier pour ces deux espèces. Ces analyses fonctionnent donc comme des tests indépendants de l’hypothèse d’une ascendance commune être humain-chimpanzé et lui fournissent des éléments de preuve indépendants.
Similarités de séquence génique chez les primates : piste de l’homologie
On définit l’homologie comme les similarités dérivées d’une ascendance commune. On sait de longue date que les êtres humains et les chimpanzés possèdent des séquences quasi-identiques pour des gènes individuels.5 Le séquençage du génome complet confirme que cette tendance à être presque identique se maintient sur la totalité du génome des deux espèces. Le génome humain comporte approximativement 3,0 x 109 nucléotides, parmi lesquels 2,7 x 109 nucléotides correspondent au génome du chimpanzé avec une différence de seulement 1,23% entre les espèces.6
En bref, la grande majorité du génome humain correspond au génome du chimpanzé avec uniquement de rares différences. La présence de divergences d’alignement de séquences entre les deux génomes, que l’on pense être dues à des insertions ou des délétions (mutations dites « indel »), réduit l’identité des deux génomes à 95% environ.7 Si l’on restreint la comparaison aux séquences codant des protéines, la valeur s’élève à 99,4%.8 Selon toute mesure, les êtres humains et les chimpanzés possèdent des génomes hautement homologues qui peuvent aisément être interprétés comme étant des copies modifiées d’un génome ancestral original.
Usage des codons dans les gènes homologues : piste de la redondance
Le code ADN utilisé pour spécifier les acides aminés dans les protéines est basé sur des triplets de nucléotides, ou « codons ». Etant donné qu’il y a quatre nucléotides (A, C, G et T), il y a 64 (c’est-à-dire 43) triplets possibles de nucléotides à disposition ; cependant, les protéines biologiques ne contiennent que vingt acides aminés. Etant donné que trois des 64 codons sont utilisés comme codons « stop » pour arrêter le processus de traduction, 61 codons sont disponibles pour encoder vingt acides aminés. Il résulte donc que la plupart des acides aminés peuvent être encodés par plus d’un codon (c’est-à-dire que le code des codons est partiellement redondant). Le schéma 1 présente une comparaison des séquences de nucléotides et d’acides aminés pour l’insuline (une hormone peptidique) chez l’être humain, le chimpanzé, le gorille, l’orang-outan, une espèce de chauve-souris et chez la souris en guise d’exemple.9
Schéma 1. Homologie des Nucléotides et des Acides Aminés de l’Insuline chez les Mammifères
[voir pages suivantes]

Schéma 1A. Séquence codante de nucléotides complète pour la pré-proinsuline alignée pour quatre espèces de primates (HS = Homo sapiens / être humain, PT = Pan troglodytes / chimpanzé, GG = Gorilla gorilla / gorille, PP = Pongo pygmaeus / orang-outan bornéen), un chiroptère (RF = Rhinolophus ferrumequinum / chauve-souris Grand Rhinolophe) et un muridé (MM = Mus musculus / souris). Les nucléotides différant de la séquence humaine sont surlignés en noir. Les acides aminés conservés chez les six espèces sont présentés sous la séquence de nucléotides. Les chiffres sous les codons conservés chez les six espèces indiquent le nombre de codons alternatifs pour cette position.

Schéma 1B. Séquence complète d’acides aminés de la pré-proinsuline alignée pour les mêmes espèces qu’en (A). Les acides aminés différant de la séquence humaine sont surlignés en noir.
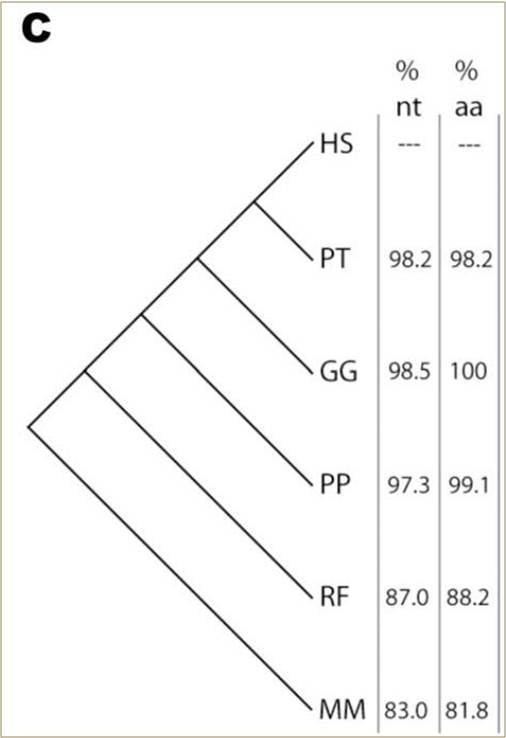
Schéma 1C. Phytogénie des mêmes six espèces, avec le pourcentage d’homologie de la pré-proinsuline par rapport à la séquence humaine présentée pour les séquences de nucléotides (nt) et d’acides aminés (aa).
Le peptide de l’insuline non-transformé chez les six espèces contient 110 acides aminés, dont la plupart peuvent être encodés par des codons alternatifs. En raison de cette redondance dans le code, il existe plus de 1019 séquences différentes de nucléotides possibles pour l’insuline humaine qui produisent toutes la même séquence observée d’acides aminés. Cependant, la séquence que l’on observe est presque identique aux séquences de nucléotides détectées chez les autres mammifères (schéma 1A). La séquence du chimpanzé diffère de seulement six nucléotides ; celle du gorille, de quatre uniquement. Au niveau de la protéine, le chimpanzé diffère de deux acides aminés, tandis que la séquence chez le gorille est identique à la nôtre (schéma 1B). Les homologies d’acides aminés et de nucléotides chez les autres mammifères s’éloignent progressivement de la séquence humaine selon un schéma niché qui suit leur phytogénie selon des critères morphologiques (schéma 1C). Bien qu’il s’agisse d’un très petit échantillon (330 nucléotides), cette tendance est représentative : une comparaison sur le génome complet des séquences codantes de l’être humain et du chimpanzé révèle qu’elles sont identiques à 99,4% sur 1,85 x 107 nucléotides.10
On peut élargir cet argument aux situations où les différences d’acides aminés sont observées pour des protéines spécifiques entre les espèces. Par exemple, les différences entre l’insuline de l’être humain et du chimpanzé au niveau de l’acide nucléique sont aussi infimes que possible malgré les différences d’acides aminés. Par exemple, chez le chimpanzé, le douzième acide aminé de l’insuline est la valine (codon CTG), alors que chez les autres mammifères que nous avons étudiés (schémas 1A, 1B), on trouve l’alanine (codons GCG ou GCC). Il y a quatre codons qui encodent la valine (GT suivi de A, C, G ou T) et quatre qui encodent l’alanine (GC suivi de A, C, G ou T). En comparant ce codon chez l’être humain et chez le chimpanzé, on trouve les deux codons les plus proches malgré la différence d’acide aminé. En d’autres termes, le code de l’acide nucléique est consistant seulement avec des changements de nucléotides uniques au sein d’une séquence ancestrale commune, en dépit des options multiples de codons pour les différents acides aminés.
On observe la même tendance en étendant ce type d’analyse à d’autres séquences d’insuline provenant d’organismes dont il est prédit qu’ils soient apparentés de manière plus éloignée à l’être humain : les gorilles et les orang-outans utilisent le même codon GCG pour l’alanine à la douzième position, tandis que les chauve-souris et les souris utilisent un codon GCC pour cette alanine. Ce schéma persiste sur toute la séquence codante de l’insuline. Une homologie significative de l’acide nucléique est maintenue malgré les options nombreuses pour la séquence conservée d’acides aminés (schéma 1C), et les changements sont hautement consistants avec des substitutions de nucléotides uniques dans la séquence ancestrale (schéma 1A). En résumé, le schéma observé d’homologie génique entre les espèces correspond précisément à ce que prédit l’ascendance commune à deux niveaux de code.
Organisation du génome dans l’espace : piste de la synténie
La synténie, dans le contexte de la génomique comparative, a trait à l’observation que les organismes apparentés exhibent non seulement une forte homologie de séquence pour des gènes individuels, mais que la disposition de ces gènes dans l’espace est similaire également. En bref, les gènes des organismes supposés de parenté évolutionnaire proche sont disposés essentiellement dans le même ordre, avec de petites différences provenant de mécanismes connus tels que les inversions de séquence, les translocations et les évènements de fusion chromosomique. Comme précédemment, l’hypothèse d’une ascendance commune prédit un tel résultat, puisque qu’on pose comme hypothèse que les deux espèces en question ne formaient antérieurement qu’une seule espèce.
On sait depuis un certain temps, grâce à des techniques de marquage de chromosomes et d’hybridation moléculaire, que les génomes de l’être humain et du chimpanzé montrent une synténie remarquable avec uniquement des différences subtiles dans l’organisation du génome.11 Neuf inversions intrachromosomiques et une fusion de chromosomes constituent les différences principales entre les séries de chromosomes de l’être humain et du chimpanzé.12 Ces observations ont maintenant été confirmées au niveau moléculaire par le séquençage du génome entier des deux espèces.13 L’exemple potentiellement le plus connu d’une différence entre les êtres humains et les chimpanzés sur le plan de l’organisation du génome est celui de la fusion télomère-télomère qui produit le chromosome 2 de l’être humain.14 Ce chromosome correspond à deux chromosomes séparés chez le chimpanzé et d’autres grands singes, ce qui suggère que le chromosome humain résulte de la fusion d’éléments qui ont persisté en tant que chromosomes distincts dans les autres espèces. Les éléments de preuve pour cette fusion proviennent de la synténie : les gènes des deux chromosomes de singe s’alignent avec ceux du chromosome humain exactement selon le schéma que produirait une fusion des deux bouts.
La synténie prédit aussi la localisation de certains produits dérivés d’une telle fusion. Les chromosomes possèdent une séquence spéciale nommée télomère à leurs bouts, ainsi qu’une séquence interne appelée centromère qui est utilisée lors de la division cellulaire. Etant donnés les deux chromosomes observés chez les singes, le chromosome 2 humain devrait comporter des séquences télomériques internes à l’endroit où son alignement passe du premier chromosome de singe au second. Il devrait aussi présenter deux centromères alignés sur les localisations correspondantes sur les chromosomes de singe. Dans les deux cas, le chromosome 2 humain est exactement conforme à ce que prédit l’ascendance commune : des séquences télomériques internes précisément au point de fusion supposé, et la présence de deux centromères aux endroits attendus, bien que l’un ait été inactivé par une accumulation de mutations.15
En résumé, lorsque l’on compare les génomes complets de l’être humain et du chimpanzé, l’on observe que la disposition des gènes dans l’espace chez les deux espèces correspond précisément à ce que l’on prédirait sur la base d’une ascendance commune : une similarité très importante, avec des différences subtiles apparaissant après la spéciation.
Archéologie génomique : piste des pseudogènes
Les pseudogènes partagés constituent un troisième élément de preuve très convaincant de l’ascendance commune des humains et des grands singes. Les pseudogènes (littéralement « faux gènes ») sont des séquences qui ont été inactivées par mutation et qui persistent dans le génome en tant que séquences non-fonctionnelles. Les pseudogènes demeurent reconnaissables pour plusieurs raisons. Premièrement, il suffit de petits changements pour inactiver un gène (par exemple, la mutation d’un seul codon en codon « stop » inapproprié tronque la traduction protéique). Dans de tels cas, les « vestiges » du gène sont presque identiques au gène fonctionnel et peuvent être identifiés aisément par leur homologie. Deuxièmement, la génomique comparative permet l’identification de pseudogènes non seulement par homologie de séquence avec des gènes fonctionnels chez d’autres organismes, mais aussi par synténie : les pseudogènes conservent leur orientation spatiale par rapport aux gènes fonctionnels adjacents après leur inactivation. Troisièmement, une fois inactivés, le rythme d’accumulation de mutations par le pseudogène n’est que lent, car les mécanismes de correction d’erreur en lecture qui régissent la réplication de l’ADN ne font pas de distinction entre les séquences d’ADN fonctionnelles et non-fonctionnelles. Ces caractéristiques permettent l’identification de pseudogènes dans des états variés de détérioration alors qu’ils mutent lentement au-delà de toute reconnaissance sur des milliers de générations.16
L’ascendance commune prédit aussi que, au-delà de l’ascendance commune être humain-chimpanzé, l’ancêtre commun des primates partage aussi une ascendance avec d’autres vertébrés dans le passé plus lointain. Par exemple, la théorie évolutionnaire prédit que les humains, comme tous les vertébrés, descendent d’ancêtres ovipares.17 Les êtres humains, comme tous les mammifères placentaires, n’utilisent pas le jaune d’oeuf comme source nutritionnelle pour leurs embryons. D’autres vertébrés tels que les poissons et les oiseaux emploient le jaune d’oeuf, tout comme un petit nombre de mammifères existants comme l’ornithorynque.
Le produit du gène vitellogenin est l’une des protéines utilisées comme constituant du jaune d’oeuf chez les vertébrés ovipares.18 Etant donné que l’on propose que les mammifères placentaires descendent d’ancêtres ovipares, des chercheurs ont récemment mené des études afin de déterminer si les vestiges de la séquence du gène vitellogenin existent chez les êtres humains sous forme de pseudogène. La localisation du gène vitellogenin fonctionnel a été déterminée dans le génome du poulet, ainsi que l’identité des gènes adjacents, afin de pouvoir situer ces gènes-là dans le génome humain. Il s’est avéré que ces gènes se trouvent côte-à-côte et sont fonctionnels dans le génome humain ; la séquence humaine les séparant a ensuite été examinée. Comme prévu, la séquence du gène vitellogenin, fortement mutée et pseudogénisée, a été identifiée dans le génome humain à cet endroit précis.19 Le génome humain contient donc les vestiges mutés d’un gène dévoué à la formation de jaune d’oeuf chez les vertébrés ovipares à l’endroit précis prédit par la synténie partagée dérivée d’une ascendance commune.
Le cas du pseudogène vitellogenin est convaincant, mais il n’est qu’un exemple parmi des milliers qui pourraient être cités.20 Par exemple, il y a des centaines de gènes utilisés pour le sens de l’odorat (gènes des récepteurs olfactifs) dans le génome humain qui sont devenus des pseudogènes.21 Par ailleurs, nombre de ces pseudogènes comportent des mutations inactivantes identiques partagées par les êtres humains, les chimpanzés et les gorilles.22 De surcroît, la détermination du degré de parenté sur la base exclusive de génomes partageant des mutations inactivantes identiques dans les pseudogènes de récepteurs olfactifs donne un classement où les humains montrent la parenté la plus proche avec les chimpanzés (plus grand nombre d’erreurs en commun), une parenté moindre avec les gorilles (moins d’erreurs en commun), et encore plus éloignée avec les orang-outans (encore moins d’erreurs en commun).23 De plus, aucun pseudogène « hors sa place » n’a été découvert dans cette étude : des pseudogènes comportant des mutations inactivantes identiques communes aux humains et aux gorilles étaient aussi présents avec des mutations identiques chez les chimpanzés ; des mutations communes aux êtres humains et aux orang-outans étaient présentes chez les chimpanzés et les gorilles.
Ce schéma correspond précisément aux prédictions de l’ascendance commune pour ces espèces, puisque la présence d’une mutation identique chez deux espèces s’explique le plus simplement par sa présence chez l’ancêtre commun des deux espèces. L’ancêtre commun des humains et des gorilles est aussi l’ancêtre commun des chimpanzés, d’où découle la prédiction que les mutations inactivantes présentes chez les humains et chez les gorilles seront aussi présentes chez les chimpanzés. En bref, l’existence de pseudogènes partagés entre les génomes des primates, leurs localisations synténiques et leurs schémas d’inactivation et de distribution soutiennent tous de manière cohérente le même modèle d’ascendance commune basé sur des critères d’homologie de séquence comparative seuls.
Génomique comparative : élément de preuve pour une ascendance commune ou conception commune ?
Lien que les éléments de preuve provenant de l’homologie, de la synténie, et de la pseudogénie soutiennent tous indépendamment l’hypothèse que les êtres humains et les chimpanzés partagent un ancêtre commun, il est aussi possible d’évaluer ces éléments d’un point de vue anti-ascendance commune tel que le dessein intelligent (DI). Il est vrai que quelques individus au sein de la mouvance DI acceptent l’ascendance commune être humain-chimpanzé,24 mais cette position n’est tenue que par ce qui semble être une minorité au sein du mouvement dans son ensemble, qui préfère une explication de conception commune plutôt que d’ascendance commune.25 La portée de cet article ne peut s’étendre à une analyse plus complète de ces questions, cependant il est instructif de considérer les éléments de preuve génomiques à la lumière d’un cadre anti-ascendance commune afin d’évaluer les forces et les faiblesses relatives du DI anti-ascendance commune et de l’ascendance commune évolutionnaire normale en tant que cadres d’explication des données de génomique comparative des primates.
Homologie, redondance et conception commune
Qu’est-ce qui empêcherait le créateur d’utiliser aussi des structures ADN et des structures corporelles similaires pour des organismes différents ?
La similarité génétique entre les chimpanzés et les êtres humains est logique d’un point de vue évolutionnaire, mais elle est aussi consistante avec le dessein intelligent.26
… les créateurs réutilisent souvent des modèles partiels pour des applications différentes. Si un créateur souhaitait générer une espèce similaire à l’être humain, il serait naturel qu’il redéploie nombre des mêmes gènes.27
Il est peut-être raisonnable de conclure qu’un créateur puisse réutiliser certaines parties pour aboutir à un produit de conception similaire (c’est-à-dire une création spéciale). Cependant, on observe que les gènes des humains et des chimpanzés se correspondent au niveau des acides aminés (c’est-à-dire au niveau fonctionnel) aussi bien qu’au niveau des codes nucléotidiques sous-jacents.
Comme nous avons vu, il existe une multitude de séquences nucléotidiques disponibles pour qu’un créateur puisse encoder une séquence d’acides aminés donnée. Même si le créateur était limité par la séquence d’acides aminés pour pouvoir générer une protéine fonctionnelle chez des organismes similaires (ce qui est douteux en soi puisque des enzymes non-homologues peuvent catalyser une réaction donnée), il lui serait aisé de choisir des codes nucléotidiques alternatifs pour éviter de donner l’apparence d’une ascendance commune. Cependant on observe, maintes et maintes fois, que les codes génétiques des organismes que l’on considère comme ayant une parenté évolutionnaire proche selon des critères non-génétiques se correspondent non seulement au niveau des acides aminés mais aussi à celui des nucléotides. C’est exactement ce que prédit l’ascendance commune, puisque l’hypothèse consiste en ce que les organismes similaires constituaient dans le passé une seule et même espèce avec un génome identique. D’un point de vue anti-ascendance commune, ce schéma est problématique.
Il suggère que le créateur ne voulait pas (ou pire, ne pouvait pas) éviter l’apparence flagrante d’une ascendance commune en mettant en oeuvre un design pour des organismes qui en réalité étaient créés séparément.
Synténie et conception commune
La littérature DI ne comporte que peu de discussions, sans grande substance, sur la synténie.
L’exemple suivant, qui tente de réfuter la conclusion que les signes de fusion chromosomique du chromosome 2 humain soutiennent l’ascendance commune, montre les arguments de base :
…les éléments de preuve provenant de la fusion chromosomique ne font que renforcer les signes d’une similarité génétique entre les chimpanzés et les êtres humains. Etant donné que la similarité était prévisible en dehors du Darwinisme et de l’ascendance commune, les similarités entre les organismes peuvent tout aussi facilement résulter des contraintes fonctionnelles mises en oeuvre par la conception commune.28
Cet argument, comme nous l’avons vu, esquive le fait que, d’un point de vue de conception commune, on ne s’attend pas nécessairement à ce que la synténie et l’homologie soient présentes ensemble.
De surcroît, la littérature DI ne mentionne pas que cette prédiction d’une « nécessité de synténie partagée » n’est pas soutenue par les éléments de preuve lorsque l’on compare les génomes d’autres groupes d’organismes hautement similaires. Par exemple, les séquences génomiques complètes de douze espèces de drosophile sont maintenant disponibles29 et leurs organisations génomiques ont été étudiées.30 Les résultats de ces analyses démontrent que le plan corporel et la biochimie de la drosophile sont bien desservis par une grande variété d’arrangements synténiques, avec des repositionnements chromosomiques beaucoup plus diversifiés dans ce groupe que dans la comparaison entre les êtres humains et les chimpanzés. En outre, la taille des gènes composant les blocs synténiques entre les espèces de drosophile est fonction du laps de temps depuis la spéciation sur la base des horloges moléculaires. Plus les séquences géniques individuelles divergent entre deux espèces, moins il y a de gènes maintenus au sein des groupements synténiques.31 Plus simplement, le créateur semble avoir employé un grand nombre d’organisations génomiques différentes pour les drosophiles, qui donnent toutes lieu à des fonctions biologiques et une morphologie de drosophile appropriées. La tendance d’une synténie décroissante correspond à celle d’une homologie de séquence génique décroissante comme prédit par la descendance commune. Il est donc plus simple d’argumenter que les différentes espèces de drosophile proviennent de modèles distincts et indépendants qu’il n’est d’argumenter que les êtres humains et les chimpanzés constituent des modèles distincts et indépendants, malgré le fait que les espèces de mouche en question sont difficiles à différencier lorsque l’on n’est pas spécialiste.
Le problème que pose cette approche argumentative DI est semblable à celui que nous avons vu avec la redondance. Il n’y a a priori aucune raison de s’attendre à un schéma d’organisation génomique similaire (c’est-à-dire une synténie partagée) entre les humains et les chimpanzés selon le raisonnement conception anti-descendance commune. De plus, on prédirait bien plutôt un schéma très différent suggérant une création spéciale et indépendante. Une fois de plus, les éléments de preuve synténiques soutiennent non seulement fortement une ascendance commune entre les humains et les chimpanzés, mais posent aussi un sérieux problème pour les interprétations anti-descendance commune.
Pseudogènes et conception commune
La littérature DI anti-descendance commune comporte trois aspects communs en ce qui concerne les pseudogènes :
1l’amalgame des pseudogènes avec l’ensemble de l’ADN non-codant sous la rubrique « ADN poubelle »
2l’absence de discussion de l’observation que les pseudogènes avec des mutations inactivantes identiques sont partagés entre les organismes exactement selon le schéma prédit par l’ascendance commune
3la suggestion que les pseudogènes possèdent une fonction qui demeure à être déterminée et qui explique leur présence découlant d’une conception délibérée.32 Le seul argument positif, celui de la fonction indéterminée des pseudogènes, ne tient pas compte des cas, nombreux, où l’on connaît la fonction d’un produit génique donné. Par exemple, la fonction du gène vitellogenin est connue, comme l’est aussi celle des nombreux récepteurs olfactifs observés en tant que pseudogènes chez les êtres humains et autres primates.
En outre, la littérature DI ne prend pas en compte l’observation que les pseudogènes sont disposés selon le modèle synténique précis que prédit l’ascendance commune. Accepter l’argument DI, c’est accepter que le créateur a placé ces séquences dans le génome humain à l’endroit synténique précis où l’on observe des versions fonctionnelles de ces gènes chez d’autres organismes, avec des séquences hautement homologues qui partagent des mutations apparentes dans une hiérarchie nichée qui correspond aux phylogénies basées sur des critères indépendants, afin d’accomplir une fonction non-apparentée qui reste à élucider. Bien qu’un telle possibilité ne puisse jamais être complètement exclue,
on se demande pourquoi le créateur aurait choisi une méthode de conception qui donnerait une impression tellement forte d’ascendance commune.
La conception commune : une théorie en crise
En résumé, l’homologie, la redondance, la synténie et les pseudogènes partagés constituent des éléments de preuve génomiques indépendants qui convergent vers une conclusion unique : les êtres humains ne sont pas des créatures de novo biologiquement indépendantes, mais partagent une ascendance commune avec d’autres formes de vie. De surcroît, les tentatives d’explication de ces données génomiques par un raisonnement anti-ascendance commune, DI et conception-commune sont extrêmement tendues et sévèrement ad hoc. Alors que chaque élément de preuve est problématique individuellement d’un point de vue anti-descendance commune, conception-commune, leur effet combiné est dévastateur.
Génomique et taille des populations d’hominidés ancestrales: la question d’Adam et Eve
Alors que beaucoup d’attention a été focalisée sur les implications du projet du génome humain pour l’ascendance commune avec les autres primates, d’autres avancées dans la génomique humaine comparative éclairent d’autres aspects de notre passé biologique. Parmi ces domaines figure l’utilisation des variations génétiques humaines actuelles pour estimer la taille effective des populations humaines ancestrales à différents moments de notre histoire évolutionnaire.
Le processus d’estimation de tailles de population à partir de données génomiques comparatives est de nature quantitative,33 et est donc moins accessible à un auditoire non-spécialiste. Cependant il est possible d’apprécier ces données qualitativement aussi bien que quantitativement. Par exemple, une fraction petite mais significative du génome humain est plus similaire au génome du gorille moderne qu’à celui du chimpanzé.34 Pour cette sous-population de séquences, l’arbre de notre espèce ne correspond pas à pas l’arbre génétique (schéma 2).35
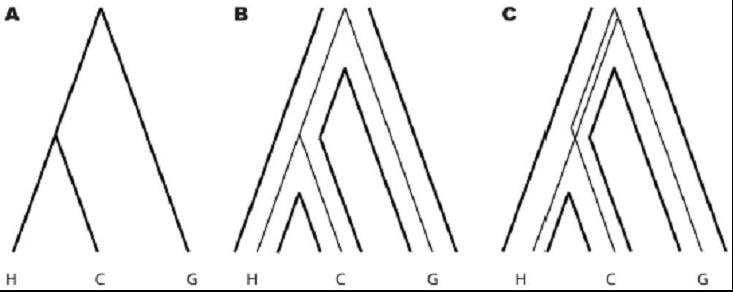
Schéma 2. Arbres de l’espèce et des gènes pour les humains, les chimpanzés et les gorilles
A. La génomique comparative des primates soutient fortement un arbre des espèces de primates regroupant les humains (H) et les chimpanzés (C) comme ayant divergé plus récemment que les gorilles (G). La plupart des gènes des humains et des chimpanzés se regroupent avant le regroupement avec le gorille (B) ; cependant, une minorité se regroupe d’abord avec les gorille (C). Cet arbre génique alternatif survient lorsque des variantes de ces gènes ont été maintenus dans la population commune ancestrale d’êtres humains-chimpanzés après la divergence des gorilles (C). On peut donc déduire la taille effective de la population de la lignée menant à l’être humain du moment présent jusqu’au point de divergence d’avec les gorilles à partir de la proportion de gènes chez les humains avec un arbre génique discordant d’avec l’arbre de l’espèce. Voir le texte pour de plus amples informations.
Cette discordance est prévue pour les espèces de parenté proche qui ont divergé les unes des autres sur une période courte.36 Autrement dit, la raison pour laquelle notre génome est tellement plus similaire à celui du chimpanzé est que nous avons eu avec le chimpanzé un ancêtre commun plus récent. Malgré cela, nous conservons des régions dans notre génome qui sont plus proches du gorille. Cette situation provient du fait que la population qui a donné lieu à l’ancêtre commun humain-chimpanzé était suffisamment grande, et suffisamment diversifiée sur le plan génétique, pour transmettre cette variation à l’être humain sans la transmettre au chimpanzé. L’être humain et le chimpanzé sont donc des échantillons génomiques distincts d’une population ancestrale diversifiée. Si ce pool avait été de moindre taille, les arbres géniques humain-chimpanzé correspondraient à l’arbre des espèces dans presque tous les cas. La proportion d’arbres géniques qui ne correspondent pas à l’arbre des espèces peut dont être utilisée pour estimer la taille de la population ancestrale.37
Des études précoces, fondées sur des séries de données limitées, ont estimé de manière consistante que la taille effective de la population ancestrale d’Homo sapiens se chiffrait autour de 10 000 individus, avec une limite inférieure de l’intervalle de confiance à 90% autour de 6 000.38 Comme cette valeur est basée sur une comparaison avec les chimpanzés et/ou les gorilles, elle représente une mesure de la taille effective de la population de notre lignée depuis la spéciation d’avec les chimpanzés (il y a environ 4-6 millions d’années) ou des gorilles (il y a environ 6-9 millions d’années).39 La disponibilité du génome complet du chimpanzé, ainsi que des séquences étendues provenant du projet en cours sur le génome du gorille, ont permis d’estimer la taille des populations ancestrales avec une précision grandissante. Des études plus récentes, consistantes avec les travaux antérieurs, fournissent des estimations de l’ordre de 8 000-10 000 avec des séries de données très importantes.40
L’étude potentiellement la plus sophistiquée à ce jour utilise les séquences génomiques complètes de l’être humain et du chimpanzé pour évaluer les arbres géniques alternatifs pour les séquences in situ dans leur contexte chromosomique humain (c’est-à-dire en tenant compte de la synténie).41 Cette étude, tout en concordant avec les estimations antérieures, démontre aussi que les séquences de l’arbre alternatif (c’est-à-dire les séquences de l’être humain et du gorille qui se regroupent avant celles de l’être humain et du chimpanzé) sont groupées en petits blocs de synténie, comme prévu.42
Des avancées récentes dans l’étude de la diversité génétique uniquement au sein de notre espèce pourvoient un moyen complémentaire d’estimer la taille effective de notre population ancestrale, en utilisation des suppositions indépendantes de celles utilisées dans les approches inter-espèces ou génomiques comparatives. Le projet international HapMap représente un effort à grande échelle pour cartographier et cataloguer les polymorphismes mononucléotidiques (Single Nucleotide Polymorphisms, ci-après SNP).43 Les SNP, tout en étant semblable à toute autre source de variation génétique lorsqu’ils sont considérés individuellement, peuvent être utilisés pour estimer la dynamique des populations ancestrales lorsqu’ils sont examinés en groupes liés sur le même chromosome grâce à un effet nommé déséquilibre de liaison (DL).44
Les SNP à liaison distante se recombinent facilement pendant la méïose, ce que ne font pas les SNP à liaison proche, et ils n’ont pas tendance à être hérités ensemble. En comparant la fréquence d’allèles à SNP individuels avec leur schéma de liaison avec d’autres SNP dans la même population, on observe que de nombreuses paires de SNP sont en DL : on les trouve liés à d’autres allèles à SNP plus fréquemment qu’on ne s’y attendrait, avec une distribution aléatoire. Le fondement biologique du DL repose sur le fait que les paires de SNP sont héritées des ancêtres et disséminées à travers une population sans être désunies : les paires liées de manière proche restent ensemble plus longtemps, et les paires liées de manière plus distante se trouvent recombinées plus rapidement. On peut donc utiliser les fréquence connues de recombinaison entre les SNP et la distribution et les proportions de paires de SNP au sein d’une population pour en estimer la taille.45 Etant donné que la fréquence de recombinaison est déterminée par la distance physique entre les paires de SNP, les études de DL peuvent être utilisées pour estimer les tailles de population dans le temps d’une manière impossible avec les estimations basées sur les mutations. La sélection de marqueurs à liaison forte permet des estimations dans le passé plus éloigné, tandis que les SNP associés de manière plus distante (avec leurs taux de recombinaison d’autant plus rapides) sont utiles pour les estimations dans les temps plus rapprochés. De plus, comme il existe plusieurs milliers de paires de SNP à examiner dans le génome humain, n’importe quelle population humaine échantillonnée pourvoit une multitude de points de données pour les méthodes basées sur le DL.
Les estimations de la dynamique de population ancestrale pour différents groupes humains actuellement disponibles avec les approches SNP/DL sont beaucoup plus détaillées que les estimations basées sur les mutations. Les groupes africains possèdent une taille de population effective supérieure (environ 7 000) aux groupes non-africains (environ 3 000) sur une période d’au moins 200 000 ans.46
Cette approche, bien qu’elle repose sur des méthodes et des hypothèses indépendantes des travaux précédents, soutient néanmoins la conclusion que les êtres humains, en tant qu’espèce, descendent d’une population ancestrale d’au moins plusieurs milliers d’individus.
De manière plus importante, l’évolutilité de cette approche révèle qu’il n’y a pas de changement significatif dans la taille de la population humaine au moment où l’être humain moderne apparaît dans les fossiles (il y a environ 200 000 ans), ou à l’époque du développement culturel et religieux significatif il y a environ 50 000 ans.47
Qu’en est-il de l’Eve mitochondriale et de l’Adam au chromosome Y ?
Les données génomiques décrites ci-dessus peuvent sembler contredire l’observation que l’ADN mitochondrial humain converge sur un ancêtre commun récent (il y a environ 170 000 ans), et que les séquences du chromosome Y humain convergent également sur un ancêtre commun plus récent encore (il y a environ 50 000 ans).48 Cette apparence de conflit, alors même qu’elle est largement exploitée dans la littérature anti-évolution,49 n’est pas juste. La convergence rapide des séquences du chromosome Y et des mitochondries s’explique par le fait qu’elles sont héritées différemment de l’ADN porté par les chromosomes (autres que le chromosome Y). L’ADN mitochondrial n’est transmis que par les mères ; les chromosomes Y ne passent que de père en fils. Il en résulte que les lignées d’ADN mitochondrial se trouvent brusquement interrompues si une mère ne donne naissance qu’à des fils ; de manière similaire, les lignées de chromosomes Y sont brusquement interrompues si un père n’engendre que des filles. Dans les deux cas, cependant, les lignées d’ADN portées par les chromosomes non-Y continuent (c’est-à-dire que les pères et les mères transmettent des chromosomes à la progéniture des deux sexes).
Prenons l’exemple d’une famille étendue (schéma 3).
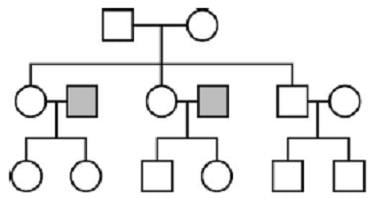
Schéma 3. Transmission mitochondriale et chromosomique chez les humains
Les carrés représentent les mâles, les cercles, les femelles. Toutes les femelles de la troisième génération ont hérité leur ADN mitochondrial de leur grand-mère commune ; cependant, elles ont hérité l’ADN chromosomique de leurs pères aussi (carrés grisés). Les variations de leur ADN chromosomique constituent donc une base appropriée pour l’estimation de la taille de leur population.
Dans cet exemple, l’ADN mitochondrial de toutes les femelles de la troisième génération provient d’un ancêtre féminin commun à la première génération. Il ressortirait de l’examination des femelles de la génération trois une convergence rapide de leur ADN mitochondrial, mais pas de leur ADN chromosomique, puisqu’il provient en partie (à 50%) de deux individus à la deuxième génération qui ne partagent pas de lien de parenté avec la source de leur ADN mitochondrial. Par conséquent, la variation de leurs séquences génomiques indiquerait qu’ils proviennent d’une population plus large qui n’a pas transmis son ADN mitochondrial à l’époque actuelle. En d’autres termes, il serait inapproprié de conclure que leur ancêtre matrilinéaire de la première génération était la seule femelle présente à l’époque, ou encore qu’elle vivait à une époque de rétrécissement significatif de la population.
Il en va de même pour les populations humaines modernes.
Malgré le fait que notre lignée d’ADN mitochondrial converge sur l’« Eve mitochondriale » dans le passé assez récent, la variation actuelle de l’ADN chromosomique humain indique qu’elle ne représentait qu’un seul individu au sein d’une population reproductrice plus large.
La même logique, mutatis mutandis, s’applique à la transmission du chromosome Y et la convergence de la variation du chromosome Y sur un « Adam » unique dans le passé récent. Bien que la convergence rapide de ces séquences d’ADN à transmission particulière soit intéressante en soi, de telles séquences ne représentent pas des mesures utiles de la taille des populations humaines ancestrales en raison de leur mode de transmission unique.50
La Genèse et le génome : « Concordisme à cliquets» ou adaptation divine ?
En résumé, les éléments de preuve génomiques donnent de deux manières l’échec à l’idée que les récits narratifs de la Genèse apportent des détails scientifiques biologiques sur l’ascendance humaine :
les êtres humains partagent une ascendance commune avec d’autres formes de vie ; et notre spéciation est le fruit non pas d’un couple ancestral mais d’une population inter-reproductrice.
De ce fait, les approches « scientifiques concordistes » à la Genèse se trouvent maintenant sous pression à la lumière de ces éléments de preuve.51 L’alliance de l’idée que la Genèse fournit des informations scientifiques – à certains niveaux au moins – à celle que la science est une initiative valide qui permet une compréhension de plus en plus fiable de la création, donne lieu à un phénomène que j’appelle le « concordisme à cliquets ». Cette approche est caractérisée par la résistance initiale de ceux qui la revendiquent aux implications de nouvelles études qui entrent en conflit avec leurs attentes concordistes, ce qui les mène fréquemment à reporter leur décision en raison de preuves insuffisantes. Cependant, si les éléments de preuve contraires à leur position continuent à s’accumuler, de tels individus peuvent à terme rejeter l’attente concordiste particulière en question, et « encliqueter » sur la position suivante qui maintient l’équilibre de leurs attentes. Au regard des éléments de preuve présentés ici, un exemple serait le décalage d’un déni de l’ascendance commune vers son acceptation, tout en retenant l’attente que notre ascendance commune dérive biologiquement d’un seul couple dans le passé récent.52
Contrairement à cette approche de concordisme à cliquets, un cadre évolutionnaire créationniste, tel que celui proposé récemment dans les travaux de Denis Lamoureux,53 accepte et incorpore aisément les données scientifiques nouvelles. Ce point de vue regarde la science dans les récits narratifs de la Genèse comme une adaptation divine à la culture de l’Ancien Proche-Orient, et ne s’attend pas à ce que la Genèse concorde avec la science moderne. Bien qu’on puisse reprocher à cette position de ne pas considérer les Ecritures avec assez de respect, le concordisme à cliquets est ouvert à la même critique, puisqu’il postule que seule une sous-partie de la Genèse contient des informations scientifiques fiables. Il en ressort donc avec cette approche que certains aspects scientifiques de la Genèse sont faussés ou obscurcis en raison d’une adaptation, alors que le livre est sensé pourvoir des informations scientifiques.
En revanche, le créationnisme évolutionnaire perçoit les récits narratifs de la Genèse comme des documents homogènes divinement adaptés à leur auditoire d’origine, des récits narratifs construits sans l’intention de répondre aux questionnements scientifiques modernes.
Article original sur le site de la fondation BioLogos
Notes
1. C. Darwin, The Descent of Man, and Selection in Relation to Sex (New York: D. Appleton and Company, 1871).
2. The Chimpanzee Sequencing and Analysis Consortium, “Initial Sequence of the Chimpanzee Genome and Comparison with the Human Genome,” Nature 437 (2005): 69–87.
3. International Human Genome Sequencing Consortium, “Initial Sequencing and Analysis of the Human Genome,” Nature 409 (2001): 860–920; International Human Genome Sequencing Consortium, “Finishing the Euchromatic Sequence of the Human Genome,” Nature 431 (2004): 931–45.
4. The Chimpanzee Sequencing and Analysis Consortium, “Initial Sequence of the Chimpanzee Genome and Comparison with the Human Genome.”
5. M. C. King and A. C. Wilson, “Evolution at Two Levels in Humans and Chimpanzees,” Science 188 (1975): 107–16.
6. The Chimpanzee Sequencing and Analysis Consortium, “Initial Sequence of the Chimpanzee Genome and Comparison with the Human Genome”; R. J. Britten, “Divergence between Samples of Chimpanzee and Human DNA Sequences Is 5%, Counting Indels,” Proceedings of the National Academy of Sciences of the USA 99 (2002): 13633–5.
7. Ibid.
8. R. Nielsen, C. Bustamante, A. G. Clark et al., “A Scan for Positively Selected Genes in the Genomes of Humans and Chimpanzees,” PLoS Biology 3 (2005): e170.
9 Les séquences d’insuline de tétrapode dans le schéma 1 ont été assemblées à partir de données recensées dans les bases de données génomiques publiques du National Center for Biotechnology Information à l’aide de recherches BLAST (http://blast.cbi.nlm.nih.gov/Blast.cgi).
10. Nielsen, Bustamante, Clark et al., “A Scan for Positively Selected Genes in the Genomes of Humans and Chimpanzees.”
11. King and Wilson, “Evolution at Two Levels in Humans and Chimpanzees”; J. W. Ijdo, A. Baldini, D. C. Ward et al., “Origin of Human Chromosome 2: An Ancestral Telomere-Telomere Fusion,” Proceedings of the National Academy of Sciences of the USA 88 (1991): 9051–5; T. Ried, N. Arnold, D. C. Ward, and J. Wienberg, “Comparative High-Resolution Mapping of Human and Primate Chromosomes by Fluorescence in Situ Hybridization,” Genomics 18 (1993): 381–6.
12. H. Kerher-Sawatzki, B. Schreiner, S. Tanzer et al., “Molecular Characterization of the Pericentric Inversion That Causes Differences between Chimpanzee Chromosome 19 and Human Chromosome 17,” American Journal of Human Genetics 71 (2002): 375–88; see also references therein.
13. L. W. Hillier, T. A. Graves, R. S. Fulton et al., “Generation and Annotation of the DNA Sequences of Human Chromosomes 2 and 4,” Nature 434 (2005): 724–31; The Chimpanzee Sequencing and Analysis Consortium, “Initial Sequence of the Chimpanzee Genome and Comparison with the Human Genome”; L. Feuk, J. R. MacDonald, T. Tang et al., “Discovery of Human Inversion Polymorphisms by Comparative Analysis of Human and Chimpanzee DNA Sequence Assemblies,” PLoS Genetics 1 (2005): e56.
14. Ijdo, Baldini, Ward et al., “Origin of Human Chromosome 2”; Hillier, Graves, Fulton et al., “Generation and Annotation of the DNA Sequences of Human Chromosomes 2 and 4”; The Chimpanzee Sequencing and Analysis Consortium, “Initial Sequence of the Chimpanzee Genome and Comparison with the Human Genome.”
15. Hillier, Graves, Fulton et al., “Generation and Annotation of the DNA Sequences of Human Chromosomes 2 and 4.”
16 Le rythme de mutation dans les pseudogènes peut sembler plus rapide que celui observé dans les séquences fonctionnelles (parce que la sélection purifiante élimine les mutations de la population), mais il est en réalité lent, au sens absolu, en raison de la correction en lecture par les ADN polymérases pendant la réplication des chromosomes.
17. D. Brawand,W. Wali, and H. Kaessmann, “Loss of Egg Yolk Genes in Mammals and the Origin of Lactation and Placentation,” PLoS Biology 6 (2006): 0507–17.
18. Ibid.
19. Ibid.
20. Cet article restreint la discussion des pseudogènes aux pseudogènes unitaires : les séquences ne possédant pas de séquence homologue au sein du même génome, mais qui sont présentes dans la région synténique attendue sous forme fonctionnelle dans d’autres organismes. En effet, si l’on considère les éléments répétitifs, les insertions endogènes rétrovirales, les pseudogènes traités, et ainsi de suite, la liste d’exemples serait multipliée maintes fois.
21. T. Olender, D. Lancet, and D. W. Nebert, “Update on the Olfactory Receptor (OR) Gene Superfamily,” Human Genomics 3 (2008): 87–97.
22. Y. Gilad,O. Man, S. Paabo, and D. Lancet, “Human Specific Loss of Olfactory Receptor Genes,” Proceedings of the National Academy of Sciences of the USA 100 (2003): 3324–7.
23. Ibid.
24. M. Behe, The Edge of Evolution: The Search for the Limits of Darwinism (New York: Free Press, 2007).
25. Par exemple, deux ouvrages grand-public DI récents tentent de jeter le doute sur l’ascendance commune humain-chimpanzé, et font l’amalgame entre l’ascendance commune et le « Darwinisme » : voir W. A. Dembski and S. McDowell, Understanding Intelligent Design: Everything You Need to Know in Plain Language (Eugene, OR: Harvest House, 2008), 55–7 and C. Luskin and L. P. Gage, “A Reply to Francis Collins’s Darwinian Arguments for Common Ancestry of Apes and Humans,” in Intelligent Design 101: Leading Experts Explain the Key Issues, ed. H. W. House (Grand Rapids, MI: Kregel Publications, 2008), 215–35.
26. Dembski and McDowell, Understanding Intelligent Design: Everything You Need to Know in Plain Language.
27. C. Luskin, “Finding Intelligent Design in Nature,” in Intelligent Design 101: Leading Experts Explain the Key Issues, 90.
28. Luskin and Gage, “A Reply to Francis Collins’s Darwinian Arguments for Common Ancestry of Apes and Humans,” 221.
29. Drosophila 12 Genomes Consortium, “Evolution of Genes and Genomes on the Drosophila Phylogeny,” Nature 450 (2007): 203-18.
30. S. W. Schaeffer, A. Bhutkar, B. F. McAllister et al., “Polytene Chromosomal Maps of 11 Drosophila Species: The Order of Genomic Scaffolds Inferred from Genetic and Physical Maps,” Genetics 179 (2008): 1601–55; A. Bhutkar, S. W. Schaeffer, S. . Russo et al., “Chromosomal Rearrangement Inferred from Comparisons of 12 Drosophila Genomes,” Genetics 179 (2008): 1657–80.
31. Ibid.
32. Pour un exemple illustrant les approches DI clé, voir Luskin and Gage, “A Reply to Francis Collins’s Darwinian Arguments for Common Ancestry of Apes and Humans,” 224–31.
33. Revu dans N. A. Rosenberg and M. Nordborg, “Genealogical Trees, Coalescent Theory and the Analysis of Genetic Polymorphisms,” Nature Reviews Genetics 3 (2002): 380–90.
34. A. Hobolth, O. F. Christensen, T. Mailund, and M. H. Schierup, “Genomic Relationships and Speciation Times of Human, Chimpanzee, and Gorilla Inferred from a Coalescent Hidden Markov Model,” PLoS Genetics 3 (2007): e7.
35. D’autres arbres géniques sont possibles, par exemple où la divergence génique se produit au sein d’une population ancestrale avant la spéciation. De plus, des facteurs additionnels tels que l’hypermutabilité doivent être pris en compte lors de l’estimation des tailles de population à partir d’arbres géniques/d’espèces discordants. Le schéma 2 est en partie adapté et résumé à partir du schéma 1 de Holboth, Christensen, Mailund et Schierup, « Genomic Relationships and Speciation Times of Human, Chimpanzee, and Gorilla Inferred from a Coaslescent Hidden Markov Model ». Se référer à l’article complet pour une discussion plus poussée ; voir aussi Rosenberg and Nordborg, “Genealogical Trees, Coalescent Theory and the Analysis of Genetic Polymorphisms.”
36. Rosenberg and Nordborg, “Genealogical Trees, Coalescent Theory and the Analysis of Genetic Polymorphisms”; Hobolth, Christensen, Mailund, and Schierup, “Genomic Relationships and Speciation Times of Human, Chimpanzee, and Gorilla Inferred from a Coalescent Hidden Markov Model.”
37. Ibid.
38.Par exemple, voir W. Li W. Li and L. A. Sadler, “Low Nucleotide Diversity in Man,” Genetics 129 (1991): 513–23.
39. Hobolth, Christensen, Mailund, and Schierup, “Genomic Relationships and Speciation Times of Human, Chimpanzee, and Gorilla Inferred from a Coalescent Hidden Markov Model.”
40. F. C. Chen and W. H. Li, “Genomic Divergences between Humans and Other Hominoids and the Effective Population Size of the Common Ancestor of Humans and Chimpanzees,” American Journal of Human Genetics 68 (2001): 444–56; Z. H. Yang, “Likelihood and Bayes Estimation of Ancestral Population Sizes in Hominoids Using Data from Multiple Loci,” Genetics 162 (2002): 1811–23; Z. Zhao, L. Jin, Y. Fu et al., “Worldwide DNA Sequence Variation in a 10-kilobase Noncoding Region on Human Chromosome 22,” Proceedings of the National Academy of Sciences of the USA 97 (2000): 11354–8.
41. Hobolth, Christensen, Mailund, and Schierup, “Genomic Relationships and Speciation Times of Human, Chimpanzee, and Gorilla Inferred from a Coalescent Hidden Markov Model.”
42. Ibid.
43. See www.hapmap.org
44. A. Tenesa, P. Navarro, B. J. Hayes et al. “Recent Human Effective Population Size Estimated from Linkage Disequilibrium,” Genome Research 17 (2007): 520–6.
45. Ibid.
46. Ibid.
47. Reasons to Believe continue à affirmer l’existence d’un Adam et d’une Eve littéraux comme progéniteurs de la race humain entière et les situe environ 50 000 ans dans le passé.
48. M. Ingman, H. Kaessmann, S. Paabo, and U. Gyllensten, “Mitochondrial Genome Variation and the Origin of Modern Humans,” Nature 408 (2000): 708–13; R. Thomson, J. K. Pritchard, P. Shen et al., “Recent Common Ancestry of Human Y Chromosomes: Evidence from DNA Sequence Data,” Proceedings of the National Academy of Sciences of the USA 97 (2000): 7360–5.
49. Par exemple, voir F. Rana and H. Ross, Who Was Adam? (Colorado Springs: Navpress, 2005), 123–31.
50. F. Ayala, A. Escalante, C. O’Huigin, and J. Klein, “Molecular Genetics of Speciation and Human Origins,” Proceedings of the National Academy of Sciences of the USA 91 (1994): 6787–94.
51. D. Lamoureux, Evolutionary Creation: A Christian Approach to Evolution (Eugene, OR: Wipf & Stock, 2008).
52. Denis Lamoureux a aussi rassemblé des éléments de preuve anecdotiques de personnes encliquetant d’une position concordiste à une autre au regard de nouvelles données (communication personnelle). De son expérience, les cliquets les plus connus sont le Créationnisme Terre Jeune, le Créationnisme Terre Ancienne, le Créationnisme Evolutionnaire qui conserve un Adam et une Eve littéraux en tant que progéniteurs biologiques de l’humanité (monogénisme évolutionnaire), et le Créationnisme Evolutionnaire proprement dit (sans attentes de concordisme scientifique avec la Genèse). D’autres gradations sont bien sûr possibles.




